Übersicht
Herkunft und Entstehungsgrund
Die Geschichte von PT-141 ist eigentlich eine Fortsetzung der Geschichte von Melanotan II. In den 1990er Jahren entwickelten Hadley und Hruby an der University of Arizona Melanotan II als Bräunungspeptid (eine Alternative zur UV-Bräunung). Die Phase-1-Studien zeigten jedoch, dass das Molekül eine starke unerwartete Nebenwirkung hatte und bei männlichen Probanden sexuelle Erregung und spontane Erektionen auslöste.
Die Forscher fragten sich: Was wäre, wenn wir diese „Nebenwirkung” als primäre Indikation isolieren würden? Sexuelle Dysfunktion ist ein riesiger therapeutischer Markt – Millionen von Menschen, besonders Frauen, leiden unter hypoaktiver sexueller Lust-Störung (HSDD), für die praktisch keine wirksame Pharmakotherapie existierte. Viagra und PDE5-Inhibitoren wirken peripher (vasodilatatorischer Effekt), was bei Männern bei erektiler Dysfunktion funktioniert, aber bei Frauen nicht wirkt, da die weibliche Sexualfunktion stärker zentral (im Gehirn) reguliert wird.
Palatin Technologies (in den 1990er Jahren speziell für die Entwicklung von Melanocortin-Agonisten gegründet) nahm Melanotan II und begann, es chemisch auf Selektivität für MC4R zu optimieren, den Rezeptor, der die zentralen sexuellen Effekte vermittelt. Nach Hunderten von Kandidatenmolekülen identifizierten sie PT-141, später als Bremelanotid benannt.
Verzweigung von Melanotan II, wichtige Verbesserungen
PT-141 und Melanotan II sind chemisch verwandt (beide sind zyklische Heptapeptide, beide mit einer Lactam-Brücke), aber mit wichtigen Unterschieden:
| Eigenschaft | PT-141 (Bremelanotid) | Melanotan II |
|---|---|---|
| Selektivität | MC4R-präferentiell (~10×) | Nicht selektiv (alle MC-Rezeptoren) |
| C-Terminus | Freies Carboxyl | Amidiert |
| Bräunungseffekt | Minimal | Stark |
| Sexueller Effekt | Stark | Stark |
| Appetitwirkung | Mild | Moderat |
| Regulatorischer Status | FDA-zugelassen (Vyleesi) | Forschungspeptid |
Praktische Konsequenz: PT-141 behält die zentralen sexuellen Effekte von Melanotan II (über MC4R), minimiert aber die kutanen und metabolischen Nebenwirkungen (über schwächere Aktivität an MC1R und MC5R).
FDA-Zulassung als Vyleesi (2019)
Nach zwei Jahrzehnten der Entwicklung und mehreren Phase-3-Studien erhielt Palatin Technologies im Juni 2019 die FDA-Zulassung für Bremelanotid unter dem Markennamen Vyleesi. Indikation: hypoaktive sexuelle Lust-Störung (HSDD) bei prämenopausalen Frauen.
Vyleesi ist ein bedeutsamer regulatorischer Meilenstein:
- Das erste von der FDA zugelassene Peptid-Sexualstimulans für Frauen
- Das zweite Medikament für weibliche HSDD (nach Addyi/Flibanserin, 2015), aber mit einem völlig anderen Mechanismus
- Ein Nachweis, dass das Melanocortin-System ein legitimes therapeutisches Ziel für sexuelle Dysfunktion ist
Vyleesi wird in einem Einweg-Autoinjektor mit einer Dosis von 1,75 mg subkutan geliefert. Es wird 45 Minuten vor der geplanten sexuellen Aktivität angewendet. Maximal 1× pro 24 Stunden, 8× pro Monat.
In der EU ist PT-141 nicht zugelassen, einige Zentren verwenden es jedoch off-label über spezielle Zugangsprogramme. Molequa® liefert PT-141 zu Forschungszwecken, für die Laborforschung und die Replikation klinischer Experimente.
Männliche Anwendung, off-label
Obwohl Vyleesi nur für Frauen zugelassen ist, wird PT-141 auch in Forschungskontexten bei Männern untersucht, insbesondere für Indikationen:
- Erektile Dysfunktion (bei der PDE5-Inhibitoren nicht gut wirken, insbesondere neurogene Formen)
- PSSD (Post-SSRI Sexual Dysfunction), persistente sexuelle Dysfunktion nach Absetzen von Antidepressiva
- Anorgasmie
- Geringe Libido bei Männern mit normalem Testosteron
Diese Anwendungen sind nicht formell zugelassen, aber mehrere klinische Studien laufen.
Wirkmechanismus, zentral, nicht peripher
Dies ist der wichtigste Aspekt von PT-141, der es von anderen Sexualstimulanzien unterscheidet.
MC4R-Aktivierung im Hypothalamus
PT-141 überquert die Blut-Hirn-Schranke und aktiviert MC4R im Hypothalamus, besonders in zwei Bereichen:
- Medialer präoptischer Bereich (MPA), das Hauptzentrum für die Integration sexuellen Verhaltens
- Paraventrikulärer Nukleus (PVN), der Koordinator neuroendokriner und autonomer Reaktionen
Die Aktivierung von MC4R in diesen Bereichen über Gαs → cAMP → PKA führt zu:
- Erhöhter dopaminerger Signalübertragung in dopaminergen Bahnen (insbesondere im mesolimbischen Pfad mit dem Belohnungssystem)
- Aktivierung oxytocinerger Neuronen
- Hemmung von Prolaktin (das ein Libido-Unterdrücker ist)
- Modulation von Serotonin in Bahnen, die die sexuelle Funktion negativ beeinflussen
Klinische Manifestation
Das Ergebnis dieser zentralen Veränderungen:
- Erhöhtes sexuelles Verlangen
- Bessere subjektive Erregung
- Bei Männern: Erektionen durch zentral aktivierte parasympathische Bahnen
- Bei Frauen: vaginale Lubrikation und klitorale Erregung über autonome Bahnen
Unterschied zu PDE5-Inhibitoren (Viagra)
Dies ist der entscheidende mechanistische Unterschied:
| Aspekt | PT-141 | PDE5-Inhibitoren (Viagra) |
|---|---|---|
| Zielort | MC4R im ZNS | PDE5 in glatter Muskulatur |
| Mechanismus | Zentral, dopaminerge Signalübertragung | Peripher, Vasodilatation |
| Effekt ohne sexuelle Stimulation | Ja, wirkt auf Verlangen | Nein, erfordert Stimulation |
| Wirkt bei Frauen | Ja, zugelassen für HSDD | Nicht effektiv |
| Wirkt bei psychogener Dysfunktion | Ja | Oft nicht |
| Unabhängig von Testosteron | Ja | Ja |
PT-141 wirkt also auf der Ebene des Verlangens und der Erregung im Gehirn, während Viagra auf der Ebene der vaskulären Reaktion in den Genitalien wirkt. Dies sind zwei völlig unterschiedliche Mechanismen, und in der klinischen Praxis können sie sich ergänzen.
Untersuchte Anwendungen
In der veröffentlichten präklinischen und klinischen Literatur werden Wirkungen von PT-141 in den folgenden Bereichen dokumentiert:
- Hypoaktive sexuelle Lust-Störung (HSDD) bei Frauen, die zugelassene Indikation (Vyleesi, 2019)
- Erektile Dysfunktion bei Männern, Phase-2-Studien (erreichten keine Phase-3-Zulassung)
- PSSD (Post-SSRI Sexual Dysfunction), sich entwickelnde Forschung in der Indikation persistenter sexueller Dysfunktion nach Antidepressiva
- Sexuelle Dysfunktion bei Diabetes, präklinische und Phase-2-Daten
- Sexuelle Dysfunktion bei Multipler Sklerose, präklinisch
- Anorgasmie, explorative klinische Erfahrungen
- Hämorrhagischer Schock, präklinische Modelle (über MC4R-vermittelten kardiovaskulären Effekt)
- Reaktive Hyperämie, präklinisch
Wissenschaft & Studien
4.1 Wichtige Publikationen
Kingsberg S.A., Clayton A.H., Portman D., et al. (2019). Bremelanotide for the Treatment of Hypoactive Sexual Desire Disorder: Two Randomized Phase 3 Trials. Obstet Gynecol. 134(5):899 bis 908., FDA-Registrierungsstudie.
Clayton A.H., Althof S.E., Kingsberg S., et al. (2016). Bremelanotide for female sexual dysfunctions in premenopausal women: a randomized, placebo-controlled dose-finding trial. Womens Health (Lond). 12(3):325 bis 337., Phase-2b-Dosisfindung.
Diamond L.E., Earle D.C., Heiman J.R., et al. (2006). An effect on the subjective sexual response in premenopausal women with sexual arousal disorder by bremelanotide (PT-141). J Sex Med. 3(4):628 bis 638., Frühe Phase-2-Daten.
Wessells H., Fuciarelli K., Hansen J., et al. (1998). Synthetic melanotropic peptide initiates erections in men with psychogenic erectile dysfunction. J Urol. 160(2):389 bis 393., Grundlegender Artikel zum erektogenen Mechanismus.
Pfaus J.G., Shadiack A., Van Soest T., et al. (2007). Selective facilitation of sexual solicitation in the female rat by a melanocortin receptor agonist. Proc Natl Acad Sci USA. 101(27):10201 bis 10204., Mechanismus im Tiermodell.
Simon J.A., Kingsberg S.A., Goldstein I., et al. (2018). Long-term safety and efficacy of bremelanotide for hypoactive sexual desire disorder. Obstet Gynecol. 134(5):909 bis 917., Langzeit-Sicherheit.
4.2 Detaillierte ausklappbare Studien
▸ Studie 1: Kingsberg 2019, FDA-Registrierungsstudie
Zitat: Kingsberg S.A., Clayton A.H., Portman D., et al. Bremelanotide for HSDD: Two Randomized Phase 3 Trials (RECONNECT). Obstet Gynecol. 2019;134(5):899 bis 908.
Was sie taten: Zwei parallele Phase-3-Studien (RECONNECT 301 und RECONNECT 302). n = 1.247 prämenopausale Frauen mit HSDD. Randomisiert, doppelblind, placebokontrolliert. Bremelanotid 1,75 mg SC selbst verabreicht nach Bedarf (vor der geplanten sexuellen Aktivität) vs. Placebo. Dauer: 24 Wochen. Primäre Endpunkte: Female Sexual Function Index (FSFI), Female Sexual Distress Scale (FSDS-DAO).
Was sie fanden:
- Signifikante Verbesserung des FSFI-Desire-Scores gegenüber Placebo in beiden Studien
- Signifikante Reduktion des FSDS-DAO (sexueller Distress und Belastung)
- 24,6 % Responder-Rate bei Bremelanotid vs. 17,1 % bei Placebo (statistische Signifikanz)
- Nebenwirkungen: Übelkeit (40 %), Flushing (20 %), Kopfschmerzen (11 %), Reaktion an der Injektionsstelle (12 %)
- 8,8 % brachen wegen Übelkeit ab
Warum es relevant ist: Dies war die FDA-Registrierungsstudie für Vyleesi. Nach 20 Jahren Entwicklung wies sie endlich eine klinisch relevante Wirksamkeit in einer schwierigen Indikation nach (HSDD ist notorisch schwierig für klinische Studien aufgrund hoher Placebo-Antworten und subjektiver Endpunkte). Die Zulassung im Juni 2019 war ein historischer Moment für die Melanocortin-Therapeutika.
▸ Studie 2: Clayton 2016, Phase-2b-Dosisfindung
Zitat: Clayton A.H., Althof S.E., Kingsberg S., et al. Bremelanotide for female sexual dysfunctions in premenopausal women. Womens Health (Lond). 2016;12(3):325 bis 337.
Was sie taten: n = 397 prämenopausale Frauen mit HSDD und/oder weiblicher sexueller Erregungsstörung (FSAD). Randomisierung: Bremelanotid 0,75, 1,25 oder 1,75 mg SC vs. Placebo. Dauer: 12 Wochen. Beurteilung: Sexual Encounter Profile (SEP), FSFI, FSDS-R.
Was sie fanden:
- Dosisabhängige Verbesserung der sexuellen Endpunkte
- Die 1,75-mg-Dosis hatte das beste Verhältnis von Wirksamkeit zu Nebenwirkungen, diese Dosis wurde für Phase 3 ausgewählt
- 0,75 mg waren subtherapeutisch
- Nebenwirkungen: überwiegend mild bis moderat, dosisabhängig
Warum es relevant ist: Die Studie definierte die optimale Dosis für das klinische Programm. Die Identifizierung von 1,75 mg als Sweet Spot zwischen Wirksamkeit und Verträglichkeit war kritisch für den Erfolg in Phase 3. Aus Forschungsperspektive ist es eine Referenz für Dosierungsprotokolle.
▸ Studie 3: Diamond 2006, frühe Phase 2
Zitat: Diamond L.E., Earle D.C., Heiman J.R., et al. An effect on the subjective sexual response in premenopausal women with sexual arousal disorder by bremelanotide (PT-141). J Sex Med. 2006;3(4):628 bis 638.
Was sie taten: n = 18 prämenopausale Frauen mit sexueller Erregungsstörung. Doppelblinde placebokontrollierte Studie. PT-141 20 mg intranasal (der damals getestete Verabreichungsweg) vs. Placebo. Beurteilung: subjektive sexuelle Reaktionen, physiologische Marker (vaginaler Photoplethysmograph), Sicherheit.
Was sie fanden:
- Signifikante Verbesserung des sexuellen Verlangens gegenüber Placebo
- Verbesserung der subjektiven Erregung und genitalen Kongestion
- Keine schweren Nebenwirkungen
- Leichte Übelkeit bei einigen Probandinnen
- Leichter Anstieg des Blutdrucks in den Stunden nach Verabreichung
Warum es relevant ist: Dies war eine der ersten klinischen Studien zu PT-141 bei Frauen. Sie validierte das Konzept, dass ein MC4R-Agonist bei Frauen wirksam sein kann (im Gegensatz zu Viagra). Der Verabreichungsweg wurde später von intranasal auf subkutan geändert (aus Sicherheitsgründen – intranasales PT-141 verursachte höhere Blutdruckwerte).
▸ Studie 4: Wessells 1998, grundlegende erektogene Studie
Zitat: Wessells H., Fuciarelli K., Hansen J., et al. Synthetic melanotropic peptide initiates erections in men with psychogenic erectile dysfunction: double-blind, placebo controlled crossover study. J Urol. 1998;160(2):389 bis 393.
Was sie taten: n = 10 Männer mit psychogener erektiler Dysfunktion. Doppelblinde placebokontrollierte Crossover-Studie mit Melanotan II (genauer dem Vorläufer von PT-141) 0,025 mg/kg SC vs. Placebo. Beurteilung: RigiScan-Tumeszenz, Dauer der Erektion, Nebenwirkungen.
Was sie fanden:
- Erektile Reaktion bei 80 % der Probanden in der aktiven Gruppe vs. 20 % Placebo
- Mittlere Zeit bis zur Erektion: 45 Minuten
- Mittlere Dauer der Erektion: 90 Minuten
- Subjektiv zufriedenstellende penetrative Fähigkeit bei 70 %
- Übelkeit bei 50 %, Flushing bei 30 %
Warum es relevant ist: Dies war der grundlegende Artikel für das gesamte Gebiet der Melanocortin-Erektogenese und sexuellen Stimulation. Wessells und Kollegen zeigten, dass der Melanocortin-Mechanismus über zentrale Pfade wirkt und sogar funktioniert, wenn der periphere PDE5-vermittelte Pfad (Viagra) versagt. Dies war der Funke, der zur Entwicklung von PT-141 als selektiverem Derivat führte.
▸ Studie 5: Pfaus 2007, tierischer Mechanismus
Zitat: Pfaus J.G., Shadiack A., Van Soest T., et al. Selective facilitation of sexual solicitation in the female rat by a melanocortin receptor agonist. Proc Natl Acad Sci USA. 2007;101(27):10201 bis 10204.
Was sie taten: Weibliche Ratten in verschiedenen endokrinen Zuständen (Proöstrus, Diöstrus, ovariektomiert) erhielten PT-141 oder Placebo. Beurteilung: proceptives sexuelles Verhalten (Aufforderung, Hops, Darts – das sind spezifische Bewegungen, mit denen das Weibchen Interesse am Männchen signalisiert), rezeptives Verhalten (Lordose), motorische Aktivität.
Was sie fanden:
- PT-141 erhöhte selektiv das proceptive (Aufforderungs-)Verhalten, d. h. die Initiative des Weibchens zum sexuellen Kontakt
- Das rezeptive Verhalten (Lordose, passive Rezeptivität) änderte sich nicht
- c-fos-Aktivierung im medialen präoptischen Bereich (MPA) des Hypothalamus, einem Schlüsselzentrum sexuellen Verhaltens
- Der Effekt war unabhängig vom endokrinen Status und wirkte auch bei ovariektomierten Weibchen (ohne Östrogen)
Warum es relevant ist: Dies ist eine mechanistische Studie, die erklärt, warum PT-141 bei Frauen wirkt. „Proceptives Verhalten” ist die Übersetzung des Konzepts von sexuellem Verlangen und Initiative aus einem Tiermodell – genau das, was PT-141 bei menschlichen Probandinnen stimuliert. Die zentrale Wirkung im MPA des Hypothalamus liefert die anatomische Grundlage für den klinischen Effekt.
▸ Studie 6: Simon 2018, Langzeit-Sicherheit
Zitat: Simon J.A., Kingsberg S.A., Goldstein I., et al. Long-term safety and efficacy of bremelanotide. Obstet Gynecol. 2018;134(5):909 bis 917.
Was sie taten: n = 684 Frauen, die Phase-3-Studien (RECONNECT) abgeschlossen hatten und in einer offenen Erweiterung weitermachten. PT-141 1,75 mg SC selbst verabreicht über 52 Wochen. Beurteilung: anhaltende Wirksamkeit, kumulative Sicherheit, Blutdruck, Herzfrequenz, Hautveränderungen.
Was sie fanden:
- Die Wirksamkeit blieb während 52 Wochen der Nachbeobachtung erhalten
- Keine neuen Sicherheitssignale im Vergleich zu Phase-3-Daten
- Übelkeit nahm bei wiederholter Anwendung ab (initiale Desensibilisierung)
- Leichter Anstieg des Blutdrucks nach jeder Dosis, aber kein kumulativer Effekt
- Keine Pigmentierungsveränderungen oder neue Nävi, ein wichtiger Unterschied zu Melanotan II
- Kein Fall eines kardialen ischämischen Vorfalls (die kardiovaskuläre Sorge war die wichtigste für die Regulierungsbehörden)
Warum es relevant ist: Die Studie war kritisch für die FDA-Zulassung und wies nach, dass die Langzeitanwendung sicher und klinisch wirksam ist. Das Fehlen kutaner Veränderungen (im Gegensatz zu Melanotan II) bestätigte die Vorteile der MC4R-Selektivität. Nach der Publikation genehmigte die FDA Vyleesi im Juni 2019.
▸ Studie 7: King 2016, Sicherheits-Übersichtsartikel
Zitat: King S.H., Mayorov A.V., Balse-Srinivasan P., et al. Melanocortin receptor agonists, structure-activity relationships, and applications in the treatment of obesity. Curr Top Med Chem. 2016;7(11):1098 bis 1106.
Was sie taten: Übersichtsartikel zur Pharmakologie und Sicherheit von Melanocortin-Agonisten – PT-141, Melanotan II, Setmelanotid, andere Kandidaten. Diskussion: Rezeptorselektivität, klinische Daten, Sicherheitsprofile, regulatorische Perspektiven.
Was sie fanden (Zusammenfassung):
- MC4R-selektive Agonisten (PT-141, Setmelanotid) haben ein besseres Sicherheitsprofil als nicht-selektive (Melanotan II)
- Hauptnebenwirkungen aller Melanocortin-Agonisten: Übelkeit, Flushing, leichter Anstieg des Blutdrucks
- Pigmentierungsveränderungen sind an die MC1R-Aktivität gebunden; selektive MC4R-Agonisten zeigen sie nicht
- Setmelanotid (Imcivree) wurde später von der FDA für seltene genetische Adipositas zugelassen (2020)
Warum es relevant ist: Der Übersichtskontext ermöglicht das Verständnis, warum PT-141 klinisch erfolgreicher war als Melanotan II. Selektivität ist entscheidend für die klinische Verträglichkeit in der Melanocortin-Pharmakologie. Dieses Prinzip führte zur Entwicklung noch selektiverer Moleküle (Setmelanotid bei Adipositas, weitere Generationen in der Palatin-Pipeline).
Lagerung
Lyophilisat (Trockenpulver vor der Rekonstitution)
- 2 Jahre bei −20 °C (Gefrierschrank)
- 18 Monate bei 2 bis 8 °C (Kühlschrank)
- Bis zu 30 Tage bei Raumtemperatur (bis zu 25 °C), lichtgeschützt und vor Feuchtigkeit geschützt
Nach der Rekonstitution (Peptid in Lösung mit bakteriostatischem Wasser)
- Bis zu 30 Tage bei 2 bis 8 °C, lichtgeschützt
- Die zyklische Struktur macht PT-141 in Lösung relativ stabil, stabiler als die meisten linearen Peptide
Praktische Lagerregeln
- Lassen Sie das Fläschchen auf Raumtemperatur erwärmen (15 bis 20 Min.), bevor Sie es öffnen.
- Vermeiden Sie Licht vollständig — PT-141 enthält Tryptophan und Histidin, die UV-empfindlich sind. Verwenden Sie eine dunkle Box im Kühlschrank.
- Vermeiden Sie den Kontakt mit starken Oxidationsmitteln.
- Nicht schütteln! Obwohl die zyklische Form robuster ist, kann mechanischer Stress die Konformation stören.
- Die Lösung sollte klar bis sehr leicht gelblich bleiben. Eine dunklere Färbung weist auf Oxidation hin, nicht verwenden.
Rekonstitution
3-Schritte-Visualisierung
- Rekonstituieren — bakteriostatisches Wasser entlang der Wand des Fläschchens hinzufügen
- Messen — mit dem Rechner (Abschnitt 8) das erforderliche Volumen berechnen
- Lagern — Kühlschrank 2 bis 8 °C, lichtgeschützt
Detailliertes Protokoll
Was Sie benötigen:
- Fläschchen mit PT-141 (5 mg Lyophilisat)
- 2 bis 2,5 ml bakteriostatisches Wasser (enthält 0,9 % Benzylalkohol, ein Konservierungsmittel, das bakterielles Wachstum verhindert)
- Insulinspritze 1 ml / 29G
Vorgehensweise:
- Lassen Sie das PT-141-Fläschchen Raumtemperatur erreichen (15 bis 20 Min.). Kaltes Fläschchen + warmes Wasser = Kondensation, die die Peptidstabilität stört.
- Desinfizieren Sie die Gummistopfen beider Fläschchen (Peptid + BAC-Wasser) mit einem Desinfektionstupfer (70 % Isopropylalkohol). Lassen Sie den Alkohol verdunsten.
- Ziehen Sie das erforderliche Volumen BAC-Wasser mit einer Insulinspritze auf. Der Standard für ein 5-mg-Fläschchen sind 2,5 ml → resultierende Konzentration 2 mg/ml = 2000 µg/ml. Dies liefert eine 1,75-mg-Dosis in einem Volumen von 0,875 ml, parallel zu Vyleesi.
- Injizieren Sie das Wasser langsam entlang der Wand des Fläschchens. Niemals direkt auf das Lyophilisat.
- Geben Sie dem Fläschchen 1 bis 2 Minuten Ruhe. PT-141 löst sich dank seiner kompakten zyklischen Struktur relativ schnell auf.
- Schwenken Sie das Fläschchen vorsichtig in kreisenden Bewegungen (NIEMALS schütteln!) für 30 bis 60 Sekunden, bis sich das gesamte Pulver gelöst hat. Die Lösung sollte vollkommen klar oder sehr leicht gelblich sein (aufgrund von Tryptophan).
- Lagern Sie im Kühlschrank bei 2 bis 8 °C, in einer dunklen Box.
Alternative Volumina für unterschiedliche Konzentrationen
| BAC-Wasser | Resultierende Konzentration | Verwendung |
|---|---|---|
| 1 ml | 5 mg/ml | Sehr hohe Konzentration (in der Forschung selten) |
| 2,5 ml | 2 mg/ml | Standard, parallel zur Vyleesi-Dosis (1,75 mg = 87 IE) |
| 5 ml | 1 mg/ml | Für niedrigere Dosen und Tiermodelle |
Regel: Für PT-141 empfehlen wir ein Volumen von 2,5 ml, um die klinische Vyleesi-Dosis parallel zu führen. Bei einer Konzentration von 2 mg/ml entspricht eine 1,75-mg-Dosis = 87,5 IE auf einer Insulinspritze (auf 88 IE runden).
Kombinationstipps — Häufig kombinierte Peptide und Moleküle
PT-141 wird in der Forschungsliteratur überwiegend allein verwendet (für die sexuelle Indikation), aber einige Kombinationen werden in explorativen Protokollen beschrieben.
Melanotan II, Alternative, keine Kombination
Dies ist eine Alternative, keine Kombination. Melanotan II ist ein nicht-selektiver MC-Agonist (stärkerer Bräunungseffekt, mehr Nebenwirkungen), PT-141 ist MC4R-selektiv (schwächerer Bräunungseffekt, saubererer sexueller Effekt). Für Forschung, die sich nur auf den sexuellen Pfad konzentriert, ist PT-141 das sauberere Molekül.
Melanotan I (Afamelanotid), für parallele kutane Indikation
Wenn die Forschung kontrollierte Bräunung (über Afamelanotid, einen MC1R-Agonisten) mit einem unabhängigen sexuellen Effekt (über PT-141, einen MC4R-Agonisten) kombinieren möchte, ist dies eine mechanistisch valide Kombination. Die Selektivität jedes Moleküls minimiert die gegenseitige Überlappung der Effekte.
Oxytocin, komplementäre sexuelle Achse
Oxytocin ist ein Peptid, das in parasympathischen und zentralen sozialen Pfaden wirkt. Während der sexuellen Aktivität vermittelt Oxytocin Bindung und die orgasmische Reaktion. PT-141 vermittelt das anfängliche Verlangen und die Erregung. Gemeinsam könnten sie ein breiteres Spektrum sexueller Funktionen abdecken. Eine hypothetische Forschungskombination, klinische Daten fehlen.
PDE5-Inhibitoren (Viagra, Cialis), für Männer
Bei erektiler Dysfunktion, bei der PDE5-Inhibitoren allein nicht ausreichen (zum Beispiel bei psychogener oder neurogener Ätiologie), kann die Kombination mit PT-141 sowohl die zentrale als auch die periphere Komponente abdecken. Vorsicht: Die Kombination kann vasoaktive Effekte verstärken (Blutdruckanstieg durch PT-141, Vasodilatation durch PDE5), erfordert Vorsicht bei kardiovaskulären Patienten.
Kisspeptin, hormonelle Achse
Kisspeptin reguliert GnRH und damit Testosteron/Östrogen. PT-141 wirkt unabhängig von Sexualhormonen. Für die Forschung bei Patienten mit niedriger Libido aufgrund hormoneller Ungleichgewichte kann die Kombination beide Ebenen ansprechen.
Keine Kombinationen mit Antidepressiva (SSRI/SNRI)
Bei SSRI/SNRI-induzierter sexueller Dysfunktion (PSSD-ähnlich) wird PT-141 als Monotherapie untersucht. Die Kombination mit aktiven Antidepressiva kann komplex sein — SSRIs erhöhen Serotonin, was den MC4R-Effekt schwächen kann. Dies ist ein aktives Forschungsgebiet.
Wichtige wissenschaftliche Daten und Zitate
“Bremelanotide is a synthetic cyclic heptapeptide melanocortin receptor agonist that acts on the central nervous system to increase sexual desire and was approved by the U.S. FDA in June 2019 for the treatment of acquired, generalized hypoactive sexual desire disorder in premenopausal women.”
— Kingsberg SA. et al. (2019), Obstet Gynecol 134(5) — PubMed 31599831
Statistiken aus der präklinischen Literatur
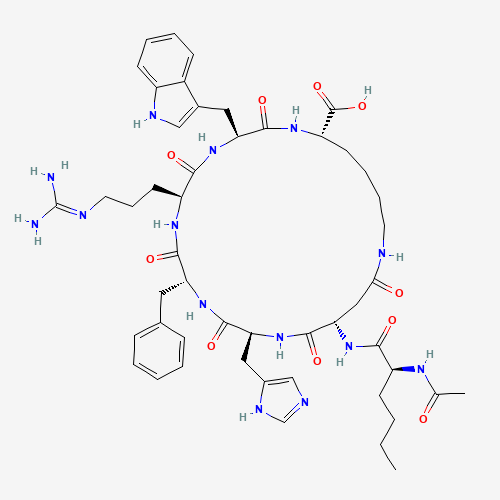
- PT-141 / Bremelanotide (Handelsname Vyleesi®), zyklisches Heptapeptid abgeleitet von Melanotan II, Sequenz Ac-Nle-cyclo(Asp-His-D-Phe-Arg-Trp-Lys)-OH, Molekulargewicht 1025,18 Da
- Entwickelt bei Palatin Technologies (USA) als Derivat des MT-II mit Selektivität für MC4R (sekundär MC1R)
- Mechanismus: zentraler MC4R-Agonist im paraventrikulären Kern des Hypothalamus, Modulation der dopaminergen Bahn (sexuelle Erregung unabhängig von der vaskulären Komponente)
- Standarddosis in klinischen Studien RECONNECT: 1,75 mg subkutan per Autoinjektor on demand, max. 1× innerhalb von 24 h, max. 8× pro Monat
- Phase 3 RECONNECT (Kingsberg 2019, n=1247 Frauen mit HSDD): ~25 % Verbesserung des FSFI-Desire-Scores vs. ~17 % Placebo (Unterschied statistisch signifikant p<0,001)
- FDA-Zulassung: 21. Juni 2019 als Vyleesi für HSDD bei prämenopausalen Frauen
- EMA: Antrag 2020 zurückgezogen (Amag Pharmaceuticals), Vyleesi ist in der EU nicht registriert
- Ca. 100+ peer-reviewed Publikationen in PubMed (2003–2024)
Referenzquellen (PubMed)
- Kingsberg SA. et al. (2019). “Bremelanotide for the Treatment of Hypoactive Sexual Desire Disorder: Two Randomized Phase 3 Trials.” Obstet Gynecol 134(5):899–908. PubMed 31599831
- Clayton AH. et al. (2016). “Bremelanotide for female sexual dysfunctions in premenopausal women: a randomized, placebo-controlled dose-finding trial.” Womens Health (Lond) 12(3):325–337. PubMed 27181790
- Diamond LE. et al. (2004). “An effect on the subjective sexual response in premenopausal women with sexual arousal disorder by bremelanotide (PT-141), a melanocortin receptor agonist.” J Sex Med 1(1):82–90. PubMed 16422988
Regulatorischer Status: Bremelanotide (PT-141) ist von der FDA seit dem 21. Juni 2019 als Vyleesi® zur Behandlung der HSDD bei prämenopausalen Frauen (Palatin/Amag/Cosette Pharmaceuticals) zugelassen. In der EU ist es nicht zugelassen (EMA-Antrag 2020 zurückgezogen). In SK/CZ/AT/PL ist es nicht registriert und nicht über die Apotheke erhältlich. Das Produkt wird ausschließlich für wissenschaftliche Laborforschung verkauft (RUO).
Häufig gestellte Fragen zu PT-141
Diese Fragen beantworten die häufigsten Suchanfragen zu PT-141 im Forschungskontext. Die vollständige technische Dokumentation finden Sie in den obigen Abschnitten.
Was ist PT-141 und wofür wird es in der Forschung verwendet?
PT-141 (Bremelanotid, 1025 Da) ist ein synthetisches zyklisches 7-Aminosäuren-Analogon des α-MSH, selektiv für die Melanocortin-Rezeptoren MC3R und MC4R. In der Forschung aktiviert es zentrale neuronale Bahnen im Hypothalamus, die die sexuelle Motivation unabhängig vom vaskulären Mechanismus (im Gegensatz zu PDE5-Inhibitoren) unterstützen. Es wurde von der FDA als Vyleesi (2019) für HSDD bei prämenopausalen Frauen zugelassen.
Welche Dosis PT-141 verwenden Wissenschaftler in Tiermodellen?
Zugelassene klinische Dosis von Vyleesi: 1,75 mg subkutan 45 Minuten vor der sexuellen Aktivität, max. 1× täglich, 8× monatlich. In präklinischen Tierstudien wurden Dosen von 0,1 bis 1 mg/kg subkutan getestet. Für die Forschung Standardkonzentration 10 mg/ml.
Was ist der Unterschied zwischen PT-141 und Melanotan II?
PT-141 ist ein selektiver MC3R/MC4R-Agonist ohne ausgeprägten MC1R-Effekt (verursacht keine Pigmentierung), während Melanotan II ein nicht-selektiver Pan-Melanocortin-Agonist ist (starke Pigmentierung + Libido + Appetitreduktion). PT-141 ist von der FDA zugelassen (Vyleesi 2019), Melanotan II nicht. PT-141 hat eine kürzere Wirkungsdauer (Stunden vs. Tage).
Ist PT-141 ein zugelassenes Arzneimittel oder eine Forschungssubstanz?
PT-141 (Bremelanotid) ist von der FDA als Vyleesi (2019) für HSDD bei prämenopausalen Frauen zugelassen, vertrieben von AMAG Pharmaceuticals/Palatin Technologies. In der EU nicht von der EMA zugelassen. Das Rohpeptid außerhalb der Vyleesi-Formulierung wird ausschließlich für die wissenschaftliche Laborforschung (RUO) verkauft.
Wie wird PT-141 gelagert und rekonstituiert?
Lyophilisiertes PT-141 bei −20 °C vor Licht geschützt lagern, Stabilität 2 bis 3 Jahre; bei 2 bis 8 °C 12 Monate. Rekonstituieren Sie mit bakteriostatischem Wasser langsam an der Innenwand der Phiole entlang, die Lösung ist 28 Tage bei 2 bis 8 °C stabil. Standardrekonstitution: 1 ml BAC-Wasser auf 10-mg-Phiole (10 mg/ml).
Wie ist die Halbwertszeit von PT-141 und wie oft wird es in Studien verabreicht?
PT-141 hat eine Plasmahalbwertszeit von ~2,7 Stunden subkutan, der biologische Effekt hält 8 bis 12 Stunden an. Im klinischen Vyleesi-Protokoll wird es 45 Minuten vor der sexuellen Aktivität verabreicht, max. einmal alle 24 Stunden und max. 8× pro Monat.
Wo kann man PT-141 in der EU für die wissenschaftliche Forschung kaufen?
PT-141 für die wissenschaftliche Forschung in der EU bietet Molequa® mit FedEx-Lieferung innerhalb von 1 bis 3 Werktagen in die Slowakei, Tschechien und die EU an. Das Produkt wird in lyophilisierter Form mit Analysenzertifikat (COA), HPLC-Reinheit ≥ 99 %, geliefert. Das Produkt ist ausschließlich für die wissenschaftliche Laborforschung bestimmt (RUO).